LA PROLIFÉRATION MONOCLONALE PLASMOCYTAIRE UN CLONE CELLULAIRE MALIN
La cellule souche proliférante du myélome multiple
Elle dériverait d’un précurseur des lymphocytes B (CD38-CD19+CD27+) qui n’exprimerait pas encore les marqueurs de la cellule myélomateuse (CD38 or CD138.
C’est une prolifération maligne à partir d’un clone unique de cellules
Ces cellules malignes synthétisent un même type d’immunoglobuline M. Ceci explique l’aspect homogène ou le pic étroit observé par électrophorèse ( DIAGNOSTIC ). L’immunoélectrophorèse montre que ces protéines appartiennent à une seule catégorie immunochimique caractérisée par la nature d es chaînes d’immunoglobulines lourdes, gamma, alpha, delta, ou epsilon, et d es chaînes d’immunoglobulines légères : kappa ou lambda.
Dans certains cas, les plasmocytes malins peuvent synthétiser une immunoglobuline complète et fonctionnelle ayant une activité anticorps normale, dans d’autres cas, les immunoglobulines sont anormales.
Les cas de proliférations biclonales, c’est-à-dire deux clones de cellules tumorales, sont très rares.
LA PROTÉINE DE BENCE-JONES
Henry Bence Jones, né en Grande Bretagne en 1813, était un chimiste devenu médecin. En 1845, à Londres, il met en évidence cette protéine chez un malade souffrant d’un myélome multiple.
Cette protéine a comme caractéristique de précipiter dans le tube à essais lorsqu’on chauffe l’échantillon d’urine à 60°C et de se dissoudre par la suite lorsque l’on continue à chauffer l’urine à 80°C, d’où le qualificatif de thermosoluble.
Cette protéine anormale est constituée de fragments d’immunoglobulines, de chaînes légères dont le faible poids moléculaire permet leur filtration par le rein et leur passage du sang vers les urines.
La protéinurie de Bence-Jones
La présence de protéines dans les urines est anormale. Ceci s’appelle une protéinurie, autrefois c’était « avoir de l’albumine dans les urines ».
La protéinurie de Bence Jones s’explique par le fait que, fréquemment, seules sont synthétisées les chaînes légères par les cellules tumorales. Dans ce cas, contrairement aux grosses protéines, ces petites protéines, de poids moléculaire inférieur à 22 000, traversent la membrane du glomérule du rein et se retrouvent dans les urines.
LA PRÉSENCE DE FACTEURS DE CROISSANCE
L’interleukine 6 (IL-6)
C’est une protéine secrétée par les lymphocytes, d’où sont nom de cytokine.
Cette cytokine est un facteur de croissance essentiel des cellules malignes du myélome multiple.
L’IL-6 est synthétisée par les cellules du micro-environnement mais également par les cellules plasmocytaires elles-mêmes.
L’IL-6 est un puissant stimulant des plasmocytes tumoraux en culture. Aux stades avancés de la maladie, les taux d’IL-6 dans le sang sont augmentés .
Les autres facteurs de croissance
Certains pourraient être impliqués dans la prolifération des plasmocytes malins. Les facteurs suivants, du moins in vitro , se comportent comme des facteurs de croissance des cellules malignes : l’IL-10, l’IL-11, le CNTF, le LIF, le G-CSF et le GM-CSF. Cela pourrait devenir des cibles intéressantes pour de futurs médicaments.
LES HYPOTHÈSES ACTUELLES
LA PHASE INFECTIEUSE : UNE STIMULATION ANTIGÈNIQUE RÉPÉTITIVE
Il semble établi que certaines mutations génétiques ont un rôle déclenchant dans le cours de la maladie.Ces différentes mutations seraient la conséquence d’un phénomène inflammatoire chronique probablement dû à une stimulation antigénique répétitive. Celle-ci pourrait être une infection virale chronique. Elle aboutirait, d’abord à une GMSI/MGUS, puis à un myélome.
LA PHASE TUMORALE : UNE SUCCESSION DE MUTATIONS
Les chercheurs ont mis en évidence récemment plusieurs mutations génétiques, caractéristiques de la maladie :
La délétion du chromosome 13q est présente dans environ 25 % des GMSI/MGUS et des myélomes indolents
La présence de mutations fréquentes du gène suppresseur de tumeur FHIT ( Fragile Histidine Triad )
Des translocations secondaires du gène c-myc
Des anomalies chromosomiques la région 14q32, qui commande la production de la protéine Bcl2, impliquée dans la survie cellulaire et dans l’inhibition de l’apoptose, pourraient expliquer l’accumulation des plasmocytes malins qui ne peuvent plus mourir
Des mutations du système Fas qui induit l’apoptose
LES ÉTAPES CONDUISANT À LA MALADIE
LA PREMIÈRE ÉTAPE : GMSI ou MGUS
L’acronyme GMSI (GaMmapathie de Signification I nconnue) ou, en anglais, MGUS ( Monoclonal Gammapathy of Uncertain Significance ) de la maladie correspondrait à la première phase de son évolution de la maladie.
Ce serait, à ce stade, l’émergence d’un clone de cellules tumorales sous l’influence soit d’un virus, encore inconnu, soit d’un état inflammatoire chronique, les deux hypothèses n’étant pas exclusives.
Ces proliférations monoclonales donnent naissance à un pic mis en évidence par l’électrophorèse des protéines qui est de faible intensité et transitoire. Elles peuvent se voir au cours de certaines infections virales et chez des patients avec un déficit immunitaire mais il ne s’agit dans aucun cas d’un myélome.
A ce stade, la maladie est totalement silencieuse et il y a ni signes cliniques ni atteinte organique. Elle est découverte, de façon fortuite, à l’occasion d’une prise de sang.
Si l’on étudie en détail le génome des plasmocytes, on s’aperçoit qu’à ce stade, la moitié des cellules ont des anomalies chromosomiques comme une translocation 14q32, impliquée dans la production des chaînes lourdes des immunoglobulines, ou une délétion du chromosome 13. L’ensemble de ces anomalies conduit à un état d’instabilité génomique.
Cet état de transition peut rester stable ou évoluer vers le stade de myélome
L’ÉVOLUTION ULTÉRIEURE : UNE SUCCESSION DE MUTATIONS…
Une première vague de mutations
L’évolution du stade de GMSI à celui de myélome, serait la conséquence de mutations successives des cellules tumorales : une translocation (4;14) associée à une dérégulation des gènes c-myc , N-ras , K-ras et Rb .
Au cours de la progression de la maladie
On assiste à une augmentation du coefficient de prolifération tumorale, une évolution plasmoblastique (forme encore plus jeune du plasmocyte).
Cette aggravation va de paire avec la présence de nouvelles anomalies génétiques comme une nouvelle translocation (16;14), une mutation du gène suppresseur de tumeur p53 .
Ces diverses mutations génétiques pourraient expliquer la stimulation de l’angiogenèse à l’origine de l’augmentation de la vascularisation de la moelle osseuse observée au cours de cette maladie et la modification du micro-environnement de la moelle osseuse.
Les mutations chromosomiques pourraient aussi expliquer l’inhibition de l’apoptose des cellules plasmocytaires malignes, en particulier en ce qui concerne la protéine Bcl2 qui a été impliquée dans la survie des cellules malignes, expliquant l’accumulation des plasmocytes tumoraux dans les organes, en particulier les os riches en moelle osseuse.
Une diminution des défenses naturelles
En parallèle, on observe une disparition de l’immunité de type cellulaire et le développement de nouvelles régulations entre les cellules, impliquant l’IL-6 et le facteur de croissance vasculaire (VEGF).
Une sidération de l’hématopoïèse normale
Elle est associée à une suppression de l’hématopoïèse normale qui se traduit par une anémie (manque de globules rouges) et une baisse du nombre de plaquettes. De plus, elle s’accompagne aussi d’une inhibition de la production de lymphocytes B normaux qui a pour conséquence, une diminution du taux des immunoglobulines et donc une susceptibilité accrue vis-à-vis des infections bactériennes.
Une augmentation de la destruction osseuse
Elle est la résultante d’une augmentation de la résorption osseuse due à la stimulation de cellules chargées du renouvellement de l’os et appelées ostéoclastes.
L’accélération de la résorption osseuse est responsable des manifestations osseuses de la maladie et de l’hypercalcémie
La prolifération tumorale plasmocytaire
Elle peut être quantifiée en mesurant la quantité d’immunoglobulines synthétisées. Des études ont permis de corréler un certain nombre de signes de la maladie à l’importance de la masse plasmocytaire. Ces études ont permis de définir des regroupements pronostiques et de mieux apprécier les effets des différents traitements.
À QUEL RYTHME PASSE-T-ON D’UN STADE A L’AUTRE ?
Le myélome multiple est une maladie dont la croissance tumorale est lente avec un temps de doublement très long. Ce temps de doublement s’échelonne sur plusieurs mois, voire des années, à sa phase initiale. En fait, seul un faible pourcentage de cellules myélomateuses est en phase S (stade de synthèse de l’ADN du cycle cellulaire). Certains myélomes de faible masse tumorale peuvent rester stables pendant des mois, voire des années, ne requérant aucun traitement spécifique. Ces formes sont parfois appelées myélomes indolents par analogie aux lymphomes non hodgkiniens dits de bas grade.
EN RÉSUMÉ …
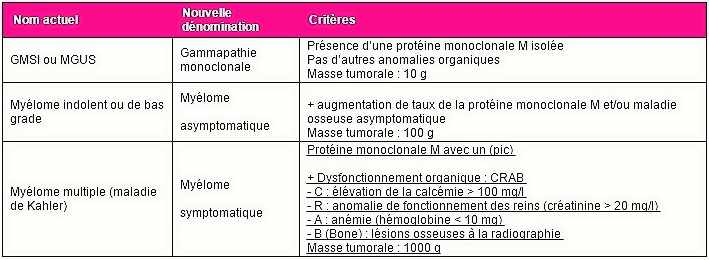
Le tableau ci-dessous résume la conception actuelle de l’évolution en trois étapes conduisant au myélome multiple ou maladie de Kahler.